Phénylcétonurie
À retenir
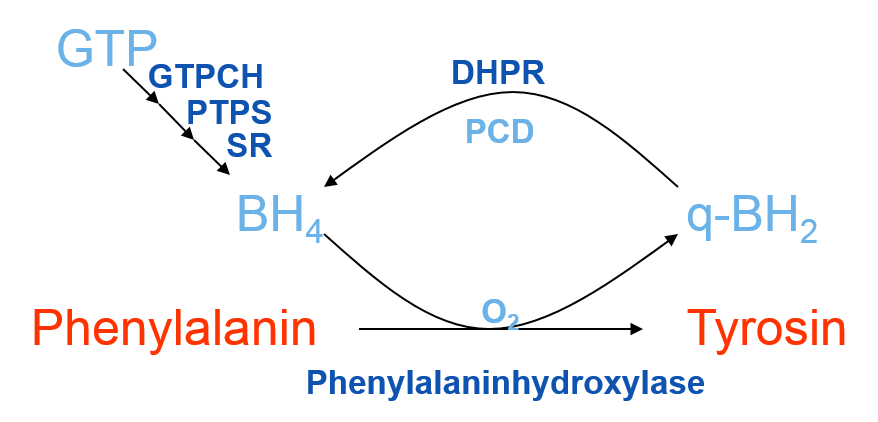
Déficit de l’enzyme phénylalanine hydroxylase qui convertit la phénylalanine en tyrosine. Maladie autosomique récessive qui, en absence de traitement, suite à l’accumulation de phénylalanine dans l’organisme entraîne une toxicité centrale causant une atteinte sévère intellectuelle, une épilepsie et des troubles du comportement.
DD: hyperphénylalaninémie suite au déficit enzymatique dans métabolisme du BH4 (pronostic moins bon)
Fait partie du dépistage néonatal en Suisse depuis 1965
Attention:…
Le suivi du régime diététique stricte est souvent difficile, surtout en âge adolescente. Des brochures d’information ont été développés pour les parents.
A l’âge adulte: problème des coûts pour aliments hypoprotéinés qui sont seulement remboursés par l’assurance invalidité (AI) jusqu’à l’âge de 20 ans.
L’association Swiss PKU offrent des informations et possibilités d’échanges avec d’autres patients.
PCU par mutations homozygotes ou hétérozygotes composées du gène codant pour phénylalanine hydroxylase ( gène PAH; OMIM *612349) sur le chromosome 12q23.2.

Incidence 1:8‘0.00 en Suisse.
Incluse dans le dépistage néonatal.
Symptômes (sans traitement):
- vers 5-6 mois: retard psychomoteur
- odeur d‘urine de souris
- eczéma
- microcéphalie
- teint clair, cheveux blonds, yeux bleus
- QI < 50
Traitement:
- régime pauvre en phénylalanine (protéine) complémenté par un mélange d’acides aminés sans phénylalanine
- traitement alternatif: traitement avec Kuvan® (BH4, le cofacteur de l’enzyme PAH), 10 mg/kg/j p.o. Seulement env. 30% des patients répondent à cette thérapie
- pendant des infections intercurrentes: ttt anti-catabolique (hydrates de carbones per os ou par sonde naso-gastrique)
En raison de la transmission autosomique récessive, un conseil génétique est recommandé (risque de récurrence: 25%).Demande des prestations médicales à l’assurance invalidité (AI).
Phénylalaninémie : le niveau recommandé varie selon l’âge (voir recommandations suisses).
surpoids (grande quantité d’hydrates de carbone)