Déficit en glutaryl-CoA déshydrogénase (DGCDH) / Acidurie glutarique type 1
À retenir
Maladie autosomique récessive caractérisée par des crises encéphalopathiques avec des lésions centrales (striatales) résultant dans un trouble neurologique irréversible avec dyskinésie dystonique.
Fait partie du dépistage néonatal en Suisse depuis 2014 (attention: patients plus âgés n'ont pas eu ce test)
Le régime strict pauvre en protéines naturelles est assez contraignant dans la vie quotidienne.
A l'âge adulte: problème des coûts pour aliments hypoprotéinés qui sont seulement remboursés par l'assurance invalidité (AI) jusqu'à l'âge de 20 ans.
Déficit en glutaryl-CoA déshydrogénase par mutations homozygotes ou hétérozygotes composées du gène codant pour glutaryl-CoA déshydrogenase (GCDH; OMIM *608801) sur le chromosome 19p13.
Incidence 1:100’000
Manifestation: lors de la première infection intercurrente (souvent entre 6 et 18 mois) par les symptômes suivants:
- perte des acquisitions
- trouble sévère neurologique avec dyskinésie dystonique (opisthotonos, dystonia, posture athetoide)
- IRM cérébrale: lésions centrales (striatales), atrophie fronto-temporale bilatérale
- développement d'une macrocéphalie
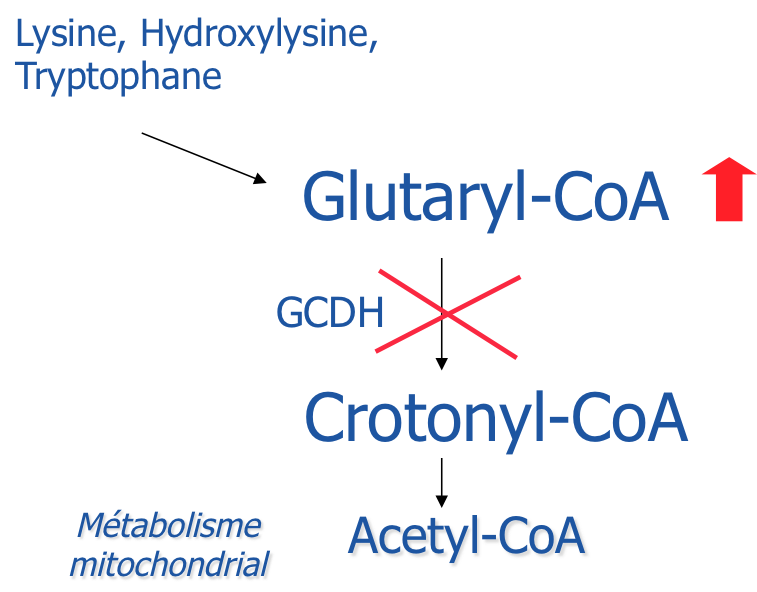
Maladie à risque de décompensation aiguë: Certificat d'urgence!
Traitement:
- régime pauvre en lysine (protéine) complémenté par un mélange d'acides aminés sans lysine et pauvre en tryptophane
- supplémentation en carnitine
- pendant des infections intercurrentes: ttt anti-catabolique (hydrates de carbones par os, par sonde naso-gastrique ou par perfusion, si nécessaire accompagné d'une petit dosage d'insuline)
Option: greffe hépatique, mais ne protège pas le cerveau!
En raison de la transmission autosomique récessive, un conseil génétique est recommandé (risque de récurrence: 25%).
Demande des prestations médicales à l'assurance invalidité (AI).
Les lésions striatales peuvent être prévenus par un traitement métabolique combiné (pas toujours).
Handicap moteur définitif après la première crise encéphalopathique