HCS (21-hydroxylase)
À retenir
Une mutation du gène de l’enzyme 21-hydoxylase est la cause la plus fréquente de HCS (hyperplasie congénitale des surrénales). Il existe 2 formes de HCS :
- forme avec perte de sel et/ou virilisante
- forme tardive ou « non-classique » toujours sans perte de sel.
La thérapie substitutive par glucocorticoïdes (Hydrocortisone) est primordiale. En cas de perte de sel, des minéralocorticoïdes et parfois du sel doivent être ajoutés.
La qualité de vie semble essentiellement normale mais dépend de la sévérité de la maladie. La possibilité d’une décompensation (crise d’Addison) est plus fréquente en bas âge.
Des associations de patients existent en Suisse comme en France
Les cellules produisant des stéroïdes (cellules des glandes surrénales et des gonades) sont d’origine mésodermique. Durant l’embryogenèse, les cellules de la surrénale migrent dans la région rétro péritonéale, où elles forment, avec des cellules du système neuronal sympathique d’origine neuro-ectodermal, la glande surrénale.
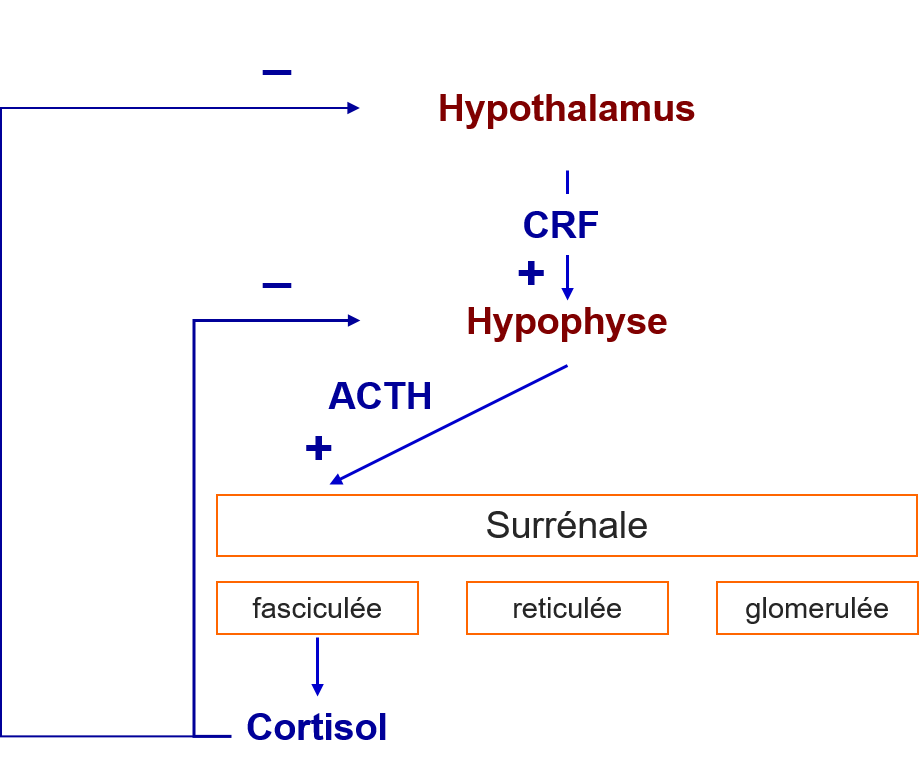
La corticosurrénale chez l’enfant et l’adulte est divisée en 3 zones et dans chacune des zones l’expression spécifique d’enzymes stéroïdiennes permettront la synthèse de certaines hormones:
- Zone glomérule —> Aldostérone (SEL)
- Zone fasciculée —> Cortisol (SUCRE)
- Zone réticulé —> Androgènes (SEX)
La synthèse de cortisol dans la zone fasciculée est principalement régulée par l’ACTH. L’ACTH (hormone hypophysaire) est elle-même régulée par CRH (croticotropin-releasing hormone) produite dans l'hypothalamus, par les mêmes neurones qui produisent la vasopressine (AVP ou ADH).
La 21-hydoxylase (P450c21) catalyse la 21-hydroxylation de progestérone en desoxycorticosterone, ainsi que l’hydoxylation de 17-OH-progestérone en 11-déoxycortisol.
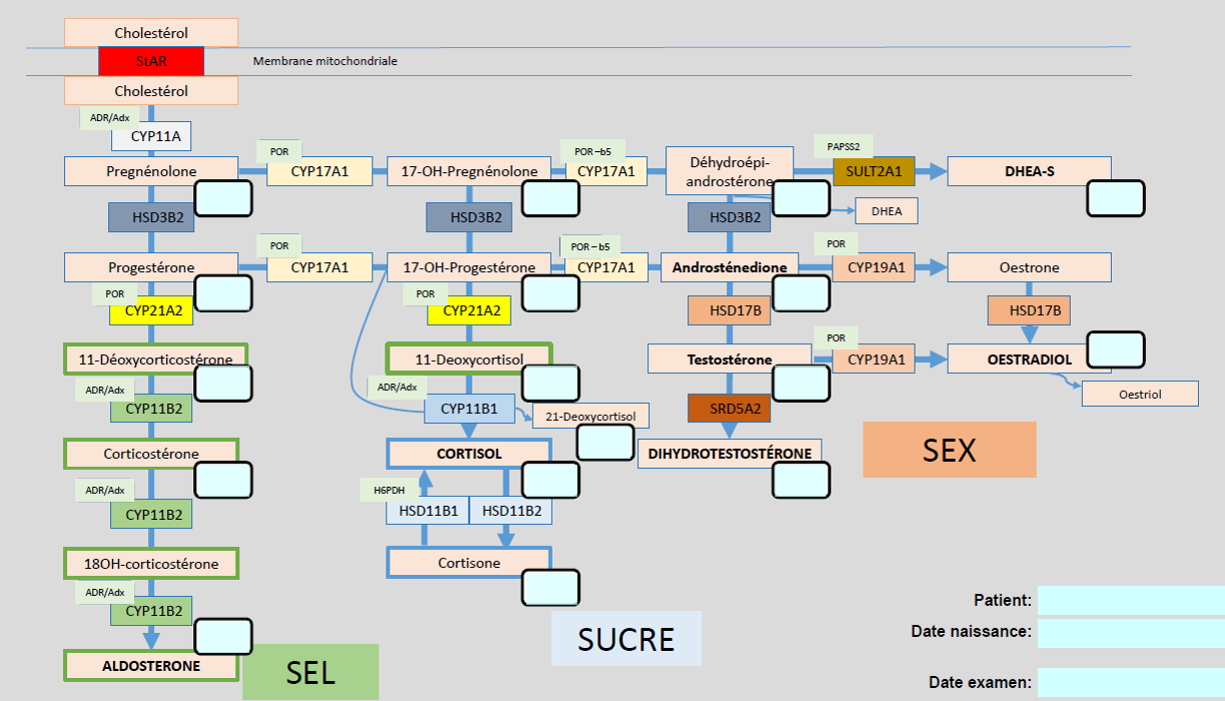
Le blocage de la 21 hydroxylase interfère autant avec la production de cortisol (SUCRE), qu'avec de l’aldostérone (SEL). L’accumulation des métaboliques avant le bloc induit une surproduction des androgènes surrénaux (SEX), pouvant masculiniser les tissus réceptifs, donc les tissus avec récepteur aux androgènes (virilisation). Le déficit de la fonction de l’enzyme 21-hydroxylase peut être complet, avec impossibilité de synthétiser le cortisol et l’aldostérone, ou le déficit peut être partiel.
Le marqueur de ce trouble est la 17-OH-progestérone. Une correlation génotype - phénotpye du CYP21 est décrite.
Sur le plan clinique, on distingue la forme « classique » avec perte de sel (déficit complet de l’activité de la 21-hydroxylase, déficit en cortisol et aldostérone) de la forme virilisante simple et la forme « non-classique » ou tardive avec hyperandrogénie mais sans déficit en cortisone sévère.La forme avec perte de sel est accompagnée d’une hyperexcrétion de sodium et d’eau ainsi que d’une réabsorption accrue du potassium.
- Doser: 17- OH-Progestérone, cortisol, ACTH, androsténedione, activité plasmatique de la rénine, aldostérone,Na, K, glycémie, gazométrie, si possible profil stéroïdien (par spectrométrie de masse (LC-MS/MS).
- Considérer: Chez l’enfant/adolescent ou adulte avec suspicion de forme tardive: test dynamique de stimulation par l’ACTH
- Interprétarion du profil stéroïdien / 17-OH-P.
- Attention: Chez le nouveau-né prématuré, d'autres normes doivent être appliqueés: LIEN: cf littérature
- Contacter endocrinologue pédiatre ou centre de référence
- Substituer avec un glucocorticoïde (Hydrocortisone), ajouter un minéralocorticoïde en cas de perte de sel.
- Prescrire une carte d’urgence avec doses de substitution glucocorticoïdes .
- Analyse génétique (gène CYP 21) : peut se faire en dehors de la phase aïgue.
La forme classique avec perte de sel est rare (1:9’000 nouveau-nés).
1:60 dans notre population est porteur hétérozygote d’une mutation du gène CYP21. Des formes plus simples dues au portage de deux formes hétérozygotes (compound hétérozygote) sont donc assez fréquentes, et l’incidence de la forme non-classique varie entre 1:53 et 1:1000 selon la population .
Il y a une corrélation génotype-(activité enzymatique)-phénotype.
- Sousdosage: Crise addisonienne (manque de cortisone aiguë), fatigue, hypoglycémie, excès androgénique avec maturation osseuse avancée et virilisation, faiblesse musculaire, perte pondérale.
- Surdosage: Hypernatriémie, hypokaliémie, hyperglycémie, rétention volumique, hypertension, ostéopénie, atrophie musculaire, signes de « Cushing »
A long terme: risque de trouble de croissance, trouble osseux et musculaire, trouble de fertilité et troubles métaboliques